The Human Genome, a Decade Later
On June 13, 2010, the New York Times ran a front-page story about the hyping of genomics. Headlined “A Decade Later, Gene Map Yields Few New Cures,” the article asserted that the Human Genome Project, the results of which were announced at a White House press conference in June 2000 and published in detail in February 2001, had yet to deliver on its promise to find the root causes of many common diseases. The genome, the story argued, held more complexity than many scientists had imagined, making it difficult to isolate the functions of the three billion DNA units, or base pairs, whose sequence the project had determined. Other journalists, and bloggers, soon weighed in somberly on the dearth of results from the project, which had taken 13 years and $3 billion to complete.

The coverage outraged Eric Lander, who was one of the leaders of the Human Genome Project and now directs the Broad Institute, a leading biomedical-research center that is a collaboration of Harvard University and MIT. “I’d like to see a quote where I ever hyped it,” says Lander. “I’m on record saying this is going to take a long time, and that the next step is to find the basis of disease, and then you have to make drugs. I said this is going to help our children’s children. Going from the germ theory of disease to antibiotics that saved people’s lives took 60 years. We might beat that. But anybody who thought in the year 2000 that we’d see cures in 2010 was smoking something.”
Lander cites a long list of technological advances and scientific insights that have come in the project’s wake. The price of sequencing DNA has dropped from hundreds of millions of dollars per person to mere thousands. The number of single-gene aberrations known to cause disease—illnesses that are invariably rare and follow a simple Mendelian pattern of inheritance—has jumped from around 100 to nearly 3,000. The growing list of common diseases that have been traced to multiple genetic variants includes everything from types of blindness to autoimmune diseases and metabolic disorders like diabetes. Studies have linked more than 200 genes to cancer—nearly three times the number that had been known of before.
Lander concedes that many features of the human genome have only recently come into clear focus—features suggesting that it’s more of a moving target than was previously thought. Genes, traditionally described as regions of DNA that code for proteins, have long been a main focus of researchers, of course. Recent studies, however, have emphasized the extraordinary power of DNA regions that do not hold the code for a protein itself but, rather, control the on/off switches that direct gene “expression,” or the extent to which that protein is actually produced. An entire world of microRNAs has moved to center stage because of their ability to silence genes.
The fledgling field of epigenetics is showing how two organisms with identical genetic sequences can have different characteristics because of heritable non-DNA factors like methyl groups, which are common reactive chemical entities that alter the behavior of genes. Many diseases have now been linked to extra or missing copies of genes, a phenomenon called copy-number variation. Researchers are also paying increased attention to transposons and other mobile genetic elements that can cause mutations, sticking themselves inside genes or deleting them altogether.
None of these factors are newly discovered, Lander stresses. But during the past 10 years, features once considered bit players have taken on the status of lead actors, now often commanding as much attention as genes themselves. So indeed, the genome contains far more inconvenient truths than was supposed a decade ago. The very idea of what we inherit and what we pass on has changed.
Yet it is “the height of silliness,” Lander contends, to suggest that this complexity makes it more difficult to develop diagnostics, treatments, and cures. He gives the example of recent studies that have linked nearly 100 genes to lipid metabolism. One of these, which has a tiny effect by itself, is the target of the statin drugs that many people take to lower cholesterol. That dozens of genes are involved in a problem, then, does not mean you need dozens of drugs to attack it; rather, he says, it reveals that there are dozens of ways to intervene. “All biological science works by collecting the complexity and recognizing it is part of a limited repertoire of events,” he says. “What’s exciting about the genome is it’s gotten us the big picture and allowed us to see the simplicity.”
The main challenge today is to catalogue and bring order to what often looks like chaos—a task that is hardly surprising to those who have worked on the genome for years. As Marc Vidal, a geneticist at the Dana-Farber Cancer Institute in Boston, says, “From the mid-1990s, there was a strong sense that just sequencing the genome wasn’t going to be enough.”
SHORTCUTS
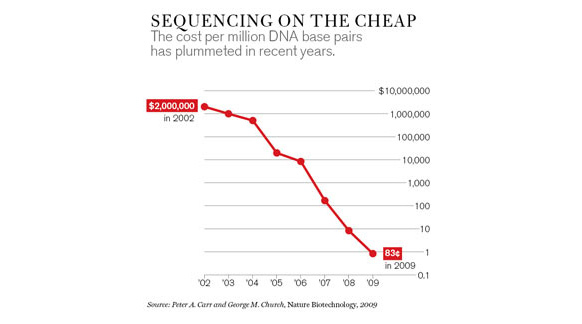
The increased understanding of the human genome has been driven largely by rapid advances in technology. And the single most profound advance has been in the cost and the speed of sequencing. “The cost dropped by a factor of 10 every year for the last five to six years, so it’s a truly amazing exponential decrease compared to the computer industry,” says Harvard University geneticist George Church, who helped develop many key sequencing technologies.
Even with the advances in technology, however, it still costs in the neighborhood of $20,000 to sequence a complete human genome—a lot less than $3 billion, but still a high price tag. And the field’s main strategy for connecting genetic makeup to observable traits—genotype to phenotype—is to compare large numbers of individuals in hopes of identifying the differences that might explain, for example, why one group develops schizophrenia and another does not.
During the past decade, genome-wide association studies, which screen many individuals in order to identify genetic variations shared by people with a particular disease, became the standard technique for comparing DNA from large groups of people without the need for complete sequences. These studies have received intense scrutiny: they were the focus of the New York Times article Landers assailed and of many critiques in the scientific literature.
The first round of these studies attempted to discover disease genes by focusing on mutations involving variations in a single DNA base pair—mutations that were relatively common, affecting more than 5 percent of the population. The idea is that these common variants, called single-nucleotide polymorphisms (SNPs), tend to occur in certain patterns within the genome and get passed down together. Thus, they serve as markers for surrounding DNA: people who share sets of SNPs, which are known as haplotype blocks, should turn out to share the same versions of specific genes. A study called the HapMap project, which looked at more than 200 people from four different populations, established a catalogue of human variation that researchers hoped would allow genome-wide association studies to find everything from genetic factors that make people sick to evolutionary traits in populations. The associations between specific haplotypes and traits did not pinpoint genetic causes but spotlighted small regions on chromosomes—regions that were thought to hold the answers.
But it turned out, as Duke University geneticist David Goldstein argued in a critique published in the April 23, 2009, issue of the New England Journal of Medicine, that “common variation is packing much less of a phenotypic punch than expected.” A study on height, Goldstein noted, had found 20 variants that together explained only about 3 percent of the variation found in humans. These sorts of results have led some researchers to scratch their heads about “missing heritability” and the “dark matter” of the genome. A key problem in using genome-wide association studies to search for the genetic roots of disease, Goldstein contended, is that many common diseases might be attributable to a number of rare variants rather than to a few common ones. He also reasoned that these studies will have little relevance in cases where large numbers of genes each make a small contribution to a disease, because “in pointing at everything, genetics would point at nothing.”
An essay in Cell a year later by Mary-Claire King and Jon McClellan of the University of Washington in Seattle fired another salvo at the genome-wide association studies. They agreed with Goldstein about the role of rare variants in disease, and they further argued that the same mutation might cause different illnesses in different people, while different mutations might all cause the same illness. Moreover, they wrote, if a mutation is so rare that it’s found only in a few individuals and their immediate families, analyzing thousands of unrelated people will miss the genetic roots of disease. They concluded that the number of genetic pathways to disease is “far greater than previously appreciated.”
Such criticisms miss the point, according to geneticist David Altshuler of Massachusetts General Hospital in Boston, who says the attacks on genome-wide association studies ignore abundant evidence that these studies have uncovered important links to disease. Some studies that tried to tie common variants to common traits found very weak associations, he concedes, but he points to several that revealed relatively large genetic factors in diabetes, heart disease in South Asians, kidney failure in African-Americans, and sickle-cell anemia in Europeans, among other illnesses.
Altshuler acknowledges that genome-wide association studies have their deficiencies, but he says that given the cost of sequencing, this shortcut has been a great boon to science. “In 10, 20 years, will you put a drop of blood in a machine and get a perfect sequence, and that’s all you do? Yes,” he says. “In 2010, is that all you should do? Not unless you’re rigid. It costs a lot more to sequence.”

Even the complete DNA sequence assembled by the Human Genome Project had a critical limitation: each individual harbors much more variation than it detected. We have a diploid genome, with one chromosome inherited from each of our parents. Thus, we have two copies each of 23 different chromosomes, but the Human Genome Project simply sequenced 23 chromosomes—a composite set assembled from several individuals. In 2007, Craig Venter, who led a private sequencing effort that was a leading competitor to the Human Genome Project, worked with Stephen Scherer, a medical geneticist at the Hospital for Sick Children in Toronto, to sequence his complete diploid genome and found more than four million differences between the chromosomes he inherited from his mother and his father. Extrapolating from this finding suggested that the amount of variation between humans was not 0.1 percent, as the Human Genome Project had estimated, but more like 0.5 percent. “For all studies, you should look at the genome in a diploid context,” says Scherer. “That’s where we need to go.”
THE CELL
Over the last decade, researchers have charted many nuanced features of the genome, and they are now fine-tuning sequencing methods and other technologies that will expand their understanding of the genomic landscape. But by and large, the dream of applying that knowledge to benefit human health remains just that. Finding usable medical information amid the huge amount of genomic data is an immense challenge.
A variety of different schemes now attempt to make sense of these mountains of data, aiming to catalogue all proteins (the proteome), RNA molecules (the transcriptome), metabolites (the metabolome), and interactions (the interactome). But some researchers argue that it’s crucial to put this vast collection of data in biological context. Sydney Brenner, a scientist at the Salk Institute for Biological Studies in La Jolla, California, and the University of Cambridge in England, takes a particularly harsh view. “This ‘omic’ science has corrupted us,” says Brenner, who won a Nobel Prize in 2002 for leading a project that four years earlier completed the first entire sequence of a multicelled organism, the worm Caenorhabditis elegans. “It has created the idea that if you just collect a lot of data, it will all work out.”
Brenner contends that the organizing principle for thinking about the genome can be found in the cell, the basic unit of life. In an essay he published in the January 12, 2010, issue of Philosophical Transactions of the Royal Society B, Brenner outlined a project called CellMap, which would catalogue every type of cell in the body and detail how different genetic regions (not genes) behave in each cellular environment. He compared it to a city map that identifies each house, the people who live inside it, and the interactions within and between the houses. “I think we should be doing genetics, not genomics,” says Brenner. “When you do genetics, you are focusing on function. When you do genomics, these are just letters and numbers. Nobody bothers about the connections.”
A cell-centric approach is only one possible route to understanding genomic data. But the conviction that DNA sequences and other genetic information won’t be medically useful until they are better connected to biology is a common one among scientists. Instead of starting with DNA and hoping to determine how it leads to complex diseases, they say, the focus needs to be on patients and what, biologically speaking, has gone wrong; then researchers can try to understand the underlying genetics. “Let’s start with the patient and work backward,” says Altshuler. “Something that has profoundly diminished the biomedical impact of [genomic] work is the unquestioned faith that everything can be learned in reductionist approaches and model systems. We need them, but we need substantial investment in studying the human being.”
Harvard’s Church agrees that not enough has been done to link genomic data to observable traits. In 2004, he launched a Personal Genome Project that ultimately aims to sequence the DNA of 100,000 people who voluntarily share their medical records and facts about their lifestyles. Without that information, understanding how an individual’s DNA sequence causes or is linked to diseases is problematic, says Church: “It’s a barrier to interpretation. It ends up oversimplifying what has to be one of the most complicated biological problems—how humans work.”
Daunting as the challenge has been to uncover the genetic basis of diseases, Altshuler says great strides have been made over the past decade. “The era of mapping genes for diseases is going to be over very soon,” he predicts. “I’m not sure whether it will be five years or 10 years—and I don’t mean we’ll explain all the heritability. But once we’ve sequenced a hundred thousand people or a million, we’ll know what there is to know.”
A decade after the completion of the Human Genome Project, scientists are still finding fundamental surprises in the way we inherit diseases (see “The Genome’s Dark Matter” ). Still, despite the unknowns, researchers are beginning to use genome data to unravel one of medicine’s greatest mysteries: how and why a cell turns cancerous (see “Cancer’s Genome”). The gap between the promise of the Human Genome Project and the realization of that promise in the clinic will surely narrow as researchers discern the complex and subtle details of the genomic landscape and the conditions that shape it. That this is taking time should come as no surprise. As Lander says, “When people say the genome is so much more complicated than we thought, you have to step back and say, ‘How simple did you think it would be?’ ”
Jon Cohen is an author and science journalist based in San Diego. His latest book is Almost Chimpanzee: Searching for What Makes Us Human, in Rainforests, Labs, Sanctuaries, and Zoos.
Keep Reading
Most Popular
Large language models can do jaw-dropping things. But nobody knows exactly why.
And that's a problem. Figuring it out is one of the biggest scientific puzzles of our time and a crucial step towards controlling more powerful future models.
How scientists traced a mysterious covid case back to six toilets
When wastewater surveillance turns into a hunt for a single infected individual, the ethics get tricky.
The problem with plug-in hybrids? Their drivers.
Plug-in hybrids are often sold as a transition to EVs, but new data from Europe shows we’re still underestimating the emissions they produce.
Stay connected
Get the latest updates from
MIT Technology Review
Discover special offers, top stories, upcoming events, and more.