The Rogue Immune Cells That Wreck the Brain
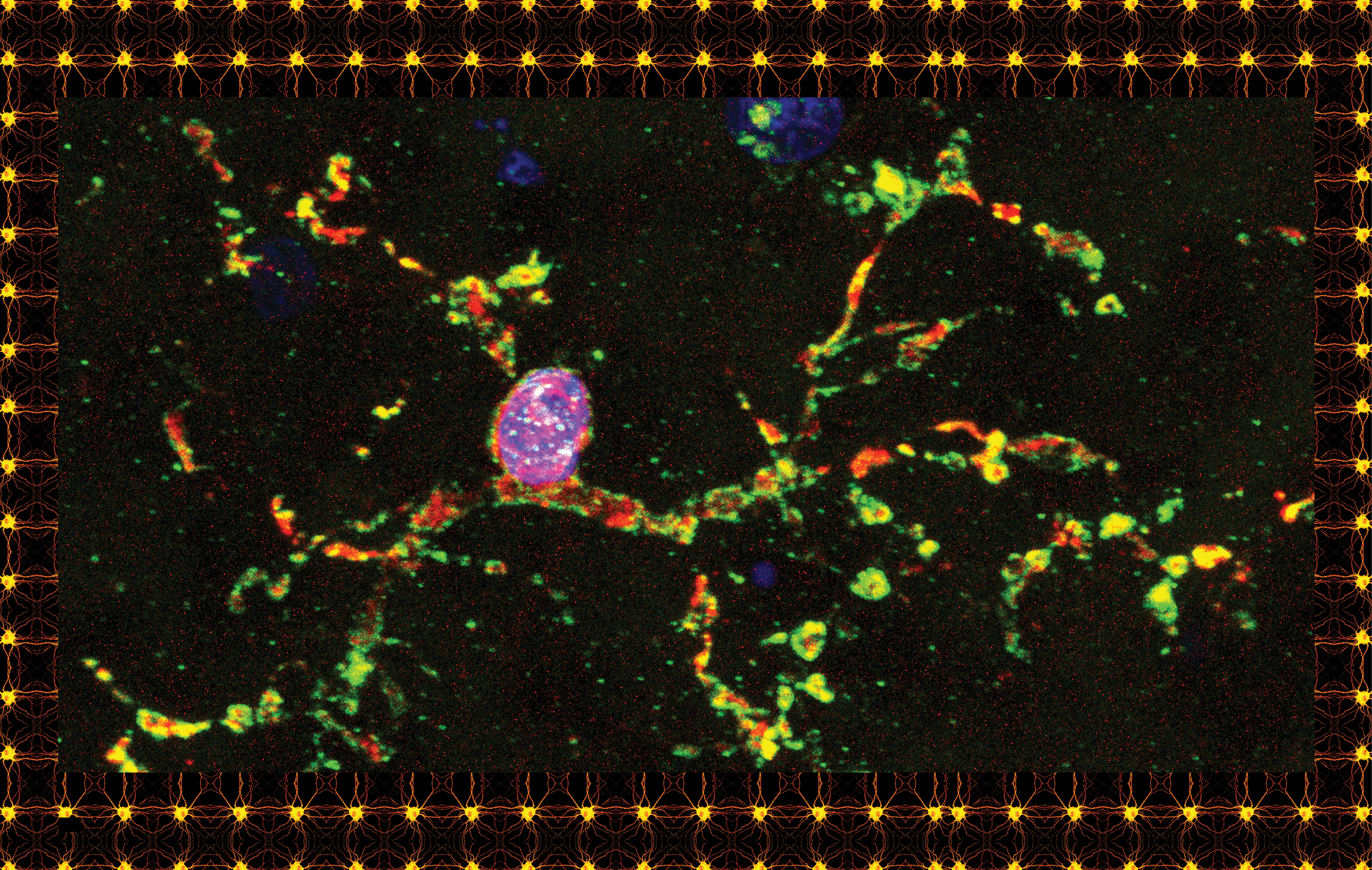
In the first years of her career in brain research, Beth Stevens thought of microglia with annoyance if she thought of them at all. When she gazed into a microscope and saw these ubiquitous cells with their spidery tentacles, she did what most neuroscientists had been doing for generations: she looked right past them and focused on the rest of the brain tissue, just as you might look through specks of dirt on a windshield.
“What are they doing there?” she thought. “They’re in the way.’”
Stevens never would have guessed that just a few years later, she would be running a laboratory at Harvard and Boston’s Children’s Hospital devoted to the study of these obscure little clumps. Or that she would be arguing in the world’s top scientific journals that microglia might hold the key to understanding not just normal brain development but also what causes Alzheimer’s, Huntington’s, autism, schizophrenia, and other intractable brain disorders.
Microglia are part of a larger class of cells—known collectively as glia—that carry out an array of functions in the brain, guiding its development and serving as its immune system by gobbling up diseased or damaged cells and carting away debris. Along with her frequent collaborator and mentor, Stanford biologist Ben Barres, and a growing cadre of other scientists, Stevens, 45, is showing that these long-overlooked cells are more than mere support workers for the neurons they surround. Her work has raised a provocative suggestion: that brain disorders could somehow be triggered by our own bodily defenses gone bad.
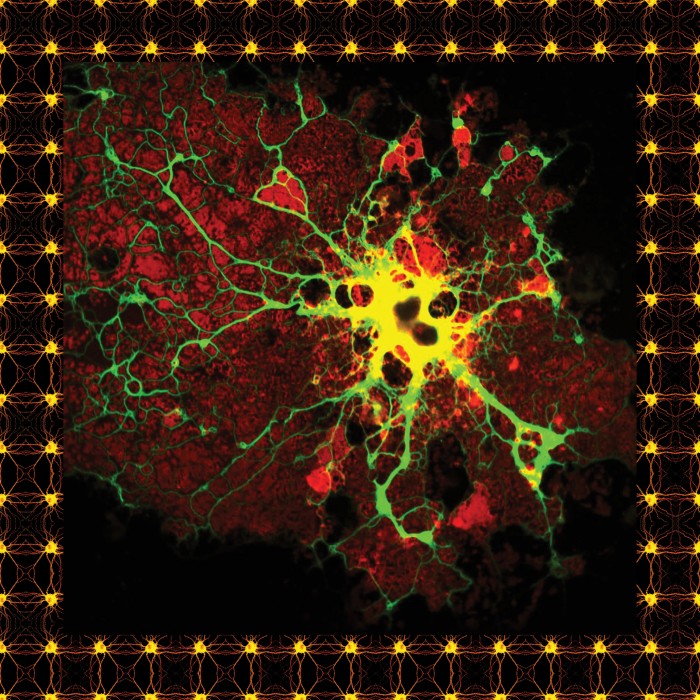
In one groundbreaking paper, in January, Stevens and researchers at the Broad Institute of MIT and Harvard showed that aberrant microglia might play a role in schizophrenia—causing or at least contributing to the massive cell loss that can leave people with devastating cognitive defects. Crucially, the researchers pointed to a chemical pathway that might be targeted to slow or stop the disease. Last week, Stevens and other researchers published a similar finding for Alzheimer’s.
This might be just the beginning. Stevens is also exploring the connection between these tiny structures and other neurological diseases—work that earned her a $625,000 MacArthur Foundation “genius” grant last September.
All of this raises intriguing questions. Is it possible that many common brain disorders, despite their wide-ranging symptoms, are caused or at least worsened by the same culprit, a component of the immune system? If so, could many of these disorders be treated in a similar way—by stopping these rogue cells?
Complex machinery
It’s not surprising that scientists for years have ignored microglia and other glial cells in favor of neurons. Neurons that fire together allow us to think, breathe, and move. We see, hear, and feel using neurons, and we form memories and associations when the connections between different neurons strengthen at the junctions between them, known as synapses. Many neuroscientists argue that neurons create our very consciousness.
Glia, on the other hand, have always been considered less important and interesting. They have pedestrian duties such as supplying nutrients and oxygen to neurons, as well as mopping up stray chemicals and carting away the garbage.
Scientists have known about glia for some time. In the 1800s, the pathologist Rudolf Virchow noted the presence of small round cells packing the spaces between neurons and named them “nervenkitt” or “neuroglia,” which can be translated as nerve putty or glue. One variety of these cells, known as astrocytes, was defined in 1893. And then in the 1920s, the Spanish scientist Pio del Río Hortega developed novel ways of staining cells taken from the brain. This led him to identify and name two more types of glial cells, including microglia, which are far smaller than the others and are characterized by their spidery shape and multiple branches. It is only when the brain is damaged in adulthood, he suggested, that microglia spring to life—rushing to the injury, where it was thought they helped clean up the area by eating damaged and dead cells. Astrocytes often appeared on the scene as well; it was thought that they created scar tissue.
This emergency convergence of microglia and astrocytes was dubbed “gliosis,” and by the time Ben Barres entered medical school in the late 1970s, it was well established as a hallmark of neurodegenerative diseases, infection, and a wide array of other medical conditions. But no one seemed to understand why it occurred. That intrigued Barres, then a neurologist in training, who saw it every time he looked under a microscope at neural tissue in distress. “It was just really fascinating,” he says. “The great mystery was: what is the point of this gliosis? Is it good? Is it bad? Is it driving the disease process, or is it trying to repair the injured brain?”
Barres began looking for the answer. He learned how to grow glial cells in a dish and apply a new recording technique to them. He could measure their electrical qualities, which determine the biochemical signaling that all brain cells use to communicate and coördinate activity.
“From the second I started recording the glial cells, I thought ‘Oh, my God!’” Barres recalls. The electrical activity was more dynamic and complex than anyone had thought. These strange electrical properties could be explained only if the glial cells were attuned to the conditions around them, and to the signals released from nearby neurons. Barres’s glial cells, in other words, had all the machinery necessary to engage in a complex dialogue with neurons, and presumably to respond to different kinds of conditions in the brain.
Why would they need this machinery, though, if they were simply involved in cleaning up dead cells? What could they possibly be doing? It turns out that in the absence of chemicals released by glia, the neurons committed the biochemical version of suicide. Barres also showed that the astrocytes appeared to play a crucial role in forming synapses, the microscopic connections between neurons that encode memory. In isolation, neurons were capable of forming the spiny appendages necessary to reach the synapses. But without astrocytes, they were incapable of connecting to one another.
Hardly anyone believed him. When he was a young faculty member at Stanford in the 1990s, one of his grant applications to the National Institutes of Health was rejected seven times. “Reviewers kept saying, ‘Nah, there’s no way glia could be doing this,’” Barres recalls. “And even after we published two papers in Science showing that [astrocytes] had profound, almost all-or-nothing effects in controlling synapses’ formation or synapse activity, I still couldn’t get funded! I think it’s still hard to get people to think about glia as doing anything active in the nervous system.”
Marked for elimination
Beth Stevens came to study glia by accident. After graduating from Northeastern University in 1993, she followed her future husband to Washington, D.C., where he had gotten work in the U.S. Senate. Stevens had been pre-med in college and hoped to work in a lab at the National Institutes of Health. But with no previous research experience, she was soundly rebuffed. So she took a job waiting tables at a Chili’s restaurant in nearby Rockville, Maryland, and showed up at NIH with her résumé every week.
After a few months, Stevens received a call from a researcher named Doug Fields, who needed help in his lab. Fields was studying the intricacies of the process in which neurons become insulated in a coating called myelin. That insulation is essential for the transmission of electrical impulses.
As Stevens spent the following years pursuing a PhD at the University of Maryland, she was intrigued by the role that glial cells played in insulating neurons. Along the way, she became familiar with other insights into glial cells that were beginning to emerge, especially from the lab of Ben Barres. Which is why, soon after completing her PhD in 2003, Stevens found herself a postdoc in Barres’s lab at Stanford, about to make a crucial discovery.
Barres’s group had begun to identify the specific compounds astrocytes secreted that seemed to cause neurons to grow synapses. And eventually, they noticed that these compounds also stimulated production of a protein called C1q.
Conventional wisdom held that C1q was activated only in sick cells—the protein marked them to be eaten up by immune cells—and only outside the brain. But Barres had found it in the brain. And it was in healthy neurons that were arguably at their most robust stage: in early development. What was the C1q protein doing there?
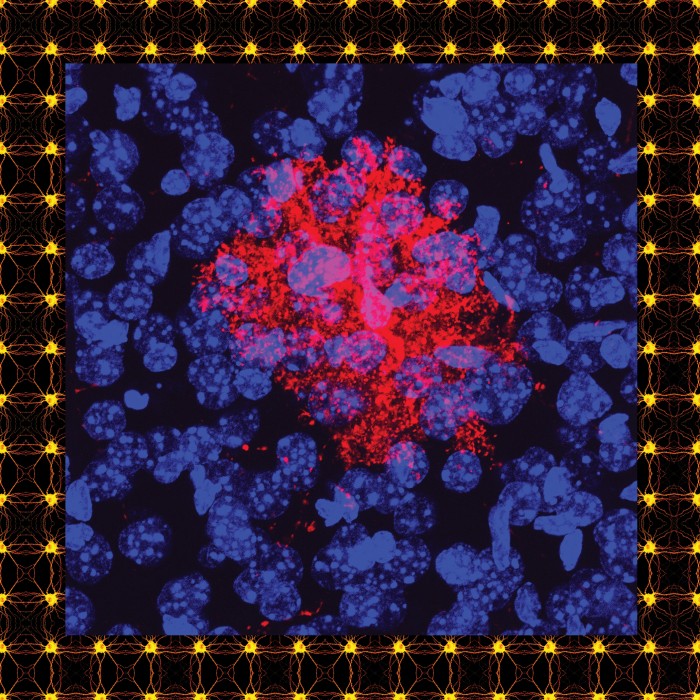
The answer lies in the fact that marking cells for elimination is not something that happens only in diseased brains; it is also essential for development. As brains develop, their neurons form far more synaptic connections than they will eventually need. Only the ones that are used are allowed to remain. This pruning allows for the most efficient flow of neural transmissions in the brain, removing noise that might muddy the signal.
But it was unknown how exactly the process worked. Was it possible that C1q helped signal the brain to prune unused synapses? Stevens focused her postdoctoral research on finding out. “We could have been completely wrong,” she recalls. “But we went for it.”
It paid off. In a 2007 paper, Barres and Stevens showed that C1q indeed plays a role in eliminating unneeded neurons in the developing brain. And they found that the protein is virtually absent in healthy adult neurons.
Now the scientists faced a new puzzle. Does C1q show up in brain diseases because the same mechanism involved in pruning a developing brain later goes awry? Indeed, evidence was already growing that one of the earliest events in neurodegenerative diseases such as Alzheimer’s, Parkinson’s, and Huntington’s was significant loss of synapses.
We might finally go after diseases that have run unchecked for generations.
When Stevens and Barres examined mice bred to develop glaucoma, a neurodegenerative disease that kills neurons in the optic system, they found that C1q appeared long before any other detectable sign that the disease was taking hold. It cropped up even before the cells started dying.
This suggested the immune cells might in fact cause the disease, or at the very least accelerate it. And that offered an intriguing possibility: that something could be made to halt the process. Barres founded a company, Annexon Biosciences, to develop drugs that could block C1q. Last week's paper published by Barres, Stevens, and other researchers shows that a compound being tested by Annexon appears to be able to prevent the onset of Alzheimer’s in mice bred to develop the disease. Now the company hopes to test it in humans in the next two years.
Paths to treatments
To better understand the process that C1q helps trigger, Stevens and Barres wanted to figure out what actually plays the role of Pac-Man, eating up the synapses marked for death. It was well known that white blood cells known as macrophages gobbled up diseased cells and foreign invaders in the rest of the body. But macrophages are not usually present in the brain. For their theory to work, there had to be some other mechanism. And further research has shown that the cells doing the eating even in healthy brains are those mysterious clusters of material that Beth Stevens, for years, had been gazing right past in the microscope—the microglia that Río Hortega identified almost 100 years ago.
Now Stevens’s lab at Harvard, which she opened in 2008, devotes half its efforts to figuring out what microglia are doing and what causes them to do it. These cells, it turns out, appear in the mouse embryo at day eight, before any other brain cell, which suggests they might help guide the rest of brain development—and could contribute to any number of neurodevelopmental diseases when they go wrong.
Meanwhile, she is also expanding her study of the way different substances determine what happens in the brain. C1q is actually just the first in a series of proteins that accumulate on synapses marked for elimination. Stevens has begun to uncover evidence that there is a wide array of protective “don’t eat me” molecules too. It’s the balance between all these cues that regulates whether microglia are summoned to destroy synapses. Problems in any one could, conceivably, mess up the system.
Evidence is now growing that microglia are involved in several neurodevelopmental and psychiatric problems. The potential link to schizophrenia that was revealed in January emerged after researchers at the Broad Institute, led by Steven McCarroll and a graduate student named Aswin Sekar, followed a trail of genetic clues that led them directly to Stevens’s work. In 2009, three consortia from around the globe had published papers comparing DNA in people with and without schizophrenia. It was Sekar who identified a possible pattern: the more a specific type of protein was present in synapses, the higher the risk of developing the disease. The protein, C4, was closely related to C1q, the one first identified in the brain by Stevens and Barres.
McCarroll knew that schizophrenia strikes in late adolescence and early adulthood, a time when brain circuits in the prefrontal cortex undergo extensive pruning. Others had found that areas of the prefrontal cortex are among those most ravaged by the disease, which leads to massive synapse loss. Could it be that over-pruning by rogue microglia is part of what causes schizophrenia?
To find out, Sekar and McCarroll got in touch with Stevens, and the two labs began to hold joint weekly meetings. They soon demonstrated that C4 also had a role in pruning synapses in the brains of young mice, suggesting that excessive levels of the protein could indeed lead to over-pruning—and to the thinning out of brain tissue that appears to occur as symptoms such as psychotic episodes grow worse.
If the brain damage seen in Parkinson’s and Alzheimer’s stems from over-pruning that might begin early in life, why don’t symptoms of those diseases show up until later? Barres thinks he knows. He notes that the brain can normally compensate for injury by rewiring itself and generating new synapses. It also contains a lot of redundancy. That would explain why patients with Parkinson’s disease don’t show discernible symptoms until they have lost 90 percent of the neurons that produce dopamine.
It also might mean that subtle symptoms could in fact be detected much earlier. Barres points to a study of nuns published in 2000. When researchers analyzed essays the nuns had written upon entering their convents decades before, they found that women who went on to develop Alzheimer’s had shown less “idea density” even in their 20s. “I think the implication of that is they could be lifelong diseases,” Barres says. “The disease process could be going on for decades and the brain is just compensating, rewiring, making new synapses.” At some point, the microglia are triggered to remove too many cells, Barres argues, and the symptoms of the disease begin to manifest fully.
Turning this insight into a treatment is far from straightforward, because much remains unclear. Perhaps an overly aggressive response from microglia is determined by some combination of genetic variants not shared by everyone. Stevens also notes that diseases like schizophrenia are not caused by one mutation; rather, a wide array of mutations with small effects cause problems when they act in concert. The genes that control the production of C4 and other immune-system proteins may be only part of the story. That may explain why not everyone who has a C4 mutation will go on to develop schizophrenia.
Nonetheless, if Barres and Stevens are right that the immune system is a common mechanism behind devastating brain disorders, that in itself is a fundamental breakthrough. Because we have not known the mechanisms that trigger such diseases, medical researchers have been able only to alleviate the symptoms rather than attack the causes. There are no drugs available to halt or even slow neurodegeneration in diseases like Alzheimer’s. Some drugs elevate neurotransmitters in ways that briefly make it easier for individuals with dementia to form new synaptic connections, but they don’t reduce the rate at which existing synapses are destroyed. Similarly, there are no treatments that tackle the causes of autism or schizophrenia. Even slowing the progress of these disorders would be a major advance. We might finally go after diseases that have run unchecked for generations.
“We’re a ways away from a cure,” Stevens says. “But we definitely have a path forward.”
Adam Piore is a freelance writer who wrote “A Shocking Way to Fix the Brain” in November/December 2015.
Keep Reading
Most Popular
Large language models can do jaw-dropping things. But nobody knows exactly why.
And that's a problem. Figuring it out is one of the biggest scientific puzzles of our time and a crucial step towards controlling more powerful future models.
How scientists traced a mysterious covid case back to six toilets
When wastewater surveillance turns into a hunt for a single infected individual, the ethics get tricky.
The problem with plug-in hybrids? Their drivers.
Plug-in hybrids are often sold as a transition to EVs, but new data from Europe shows we’re still underestimating the emissions they produce.
Google DeepMind’s new generative model makes Super Mario–like games from scratch
Genie learns how to control games by watching hours and hours of video. It could help train next-gen robots too.
Stay connected
Get the latest updates from
MIT Technology Review
Discover special offers, top stories, upcoming events, and more.