First Atomic Level Simulation of a Whole Battery
When it comes to developing the next generation of technology, the biggest bottleneck is arguably the battery. Engineers need better batteries for electric vehicles, for energy storage in power grids and, of course, for consumer electronic devices.
These batteries need to deliver a higher current over more discharge cycles with a greater energy density, to name just a few of the challenges.
Building and testing new battery designs is time-consuming, difficult and expensive. So it is handy for electrochemists to simulate the way a battery performs before they ever get their hands dirty.
That’s tricky. Nobody has been able to simulate an entire battery on the atomic level because of the complexity of the processes that go on and the limitations of today’s modelling techniques.
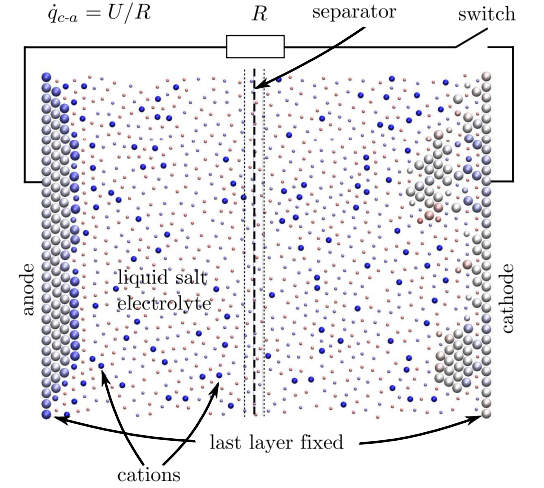
Today that changes thanks to the work of Wolf Dapp at the Institute for Advanced Simulation and Martin Muser at the University of Saarlandes, both in Germany. These guys have simulated the behaviour of a whole battery on the atomic scale. And their simulation reproduces many of the real characteristics of a battery from first principles for the first time.
In recent years, computer scientists have made significant progress in simulating various aspects of battery behaviour. These models work on the mesoscale–smaller than the electrodes but larger than the molecules. The simulations rely on experimental data to model things like ionic and electronic conductivity, diffusion coefficients, current densities, electrochemical potentials and so on.
These models have a serious drawback– they have little predictive power when it comes to new materials or combinations of materials for which experimental data is not available. To predict the behaviour of new materials, electrochemists need to model batteries on the scale of atoms and molecules.
That’s hard because the techniques computer scientists use to simulate the behaviour of atoms and molecules are not suitable for batteries. These simulations are designed for systems that are in equilibrium or close to it. They work by equalising the chemical potential or minimising the energy of the system. “However, the difference in the chemical potential between two electrodes is precisely what drives charge transport in a battery,” say Dapp and Muser.
So to model a battery as a whole, the computer model must take into account any change in energy or chemical potential at each step in the calculation. This is exactly what Dapp and Muser have done. In their model, charge is a variable that can be exchanged between atoms and across bonds at each stage of the calculation.
The resulting simulations are small but impressive. Their nanobattery consists of 358 atoms, of which 118 make up the electrodes. The cathode is initially covered with a layer of 20 atoms with 39 positively ionised atoms dissolved in the electrolyte.
The calculation then proceeds in steps in which atoms can move and exchange charge as the system evolves. The entire simulation consists of some 10 million of these steps.
The results are remarkable because they actually reproduce the generic discharge curves of real macroscopic batteries. For example, a lower operating temperature reduces the effective capacity of the simulated battery. And most important of all, the simulation reproduces the way ordinary batteries wear out. “Upon recharging, the battery performance degrades slightly, and the electrode surface morphology changes during the battery’s operation,” say Dapp and Muser.
These guys say that the work at this stage is just a proof-of-principle model and that there are various ways in which it can be improved in future. For example, they model the electrolyte using particles that have a fixed charge and so cannot exchange it.
That’s not how electrolytes work in real batteries and so this is an important shortcoming of the new method. But Dapp and Muser intend to correct this. “This idealization will be abandoned in future work,” they say.
Overall, this looks to be important work. This kind of model could dramatically improve the predictive power of battery simulations and thereby help to save electrochemists significant time, energy and money before they begin detailed experiments.
The end result should be better batteries but there is a long way to go before then.
Ref: arxiv.org/abs/1308.3424 : Redox Reactions With Empirical Potentials: Atomistic Battery Discharge Simulations
Keep Reading
Most Popular
Large language models can do jaw-dropping things. But nobody knows exactly why.
And that's a problem. Figuring it out is one of the biggest scientific puzzles of our time and a crucial step towards controlling more powerful future models.
The problem with plug-in hybrids? Their drivers.
Plug-in hybrids are often sold as a transition to EVs, but new data from Europe shows we’re still underestimating the emissions they produce.
How scientists traced a mysterious covid case back to six toilets
When wastewater surveillance turns into a hunt for a single infected individual, the ethics get tricky.
Google DeepMind’s new generative model makes Super Mario–like games from scratch
Genie learns how to control games by watching hours and hours of video. It could help train next-gen robots too.
Stay connected
Get the latest updates from
MIT Technology Review
Discover special offers, top stories, upcoming events, and more.