Measuring the shape of proteins just got easier thanks to mathematics
Proteins are the building blocks of life. They are long chains of amino acids that self-assemble into molecular machines of extraordinary complexity. These machines include computing devices called ribosomes, scaffolding skeletons called microtubules, and leg-like walking machines called kinesin, among many others.
This process of self-assembly is one of the great marvels of modern science—almost as if a chain of Lego bricks were to suddenly assemble into a robot. Nobody is quite sure how this happens, but scientists do know that the shape of the resulting structure determines the protein’s function and how it interacts with other proteins.
So measuring the shape of proteins is a crucial task. The most common method is to form crystals from proteins and then use x-ray crystallography to determine the protein structure.
That’s a problem because most proteins do not form crystals. And even when they do, the protein molecules may not all take the same shape as they become packed, leading to inaccuracies.
Another technique, called nuclear magnetic resonance, creates images of proteins in solution but requires them to be tightly packed into bundles. Again, only a few proteins can do this.
However, a tiny fraction of proteins can be imaged with both techniques. That’s handy because it allows molecular biologists to compare the structures that each technique produces.
And it turns out that the structures found by each technique differ in significant ways. But it isn’t clear exactly what causes the differences and how to interpret them.
Today that changes, at least in part, thanks to the work of Zhe Mei and colleagues at Yale University in New Haven. This team has measured the difference in the protein structure determined by x-ray crystallography and NMR. And they’ve worked out why the difference arises and how to correct for it.
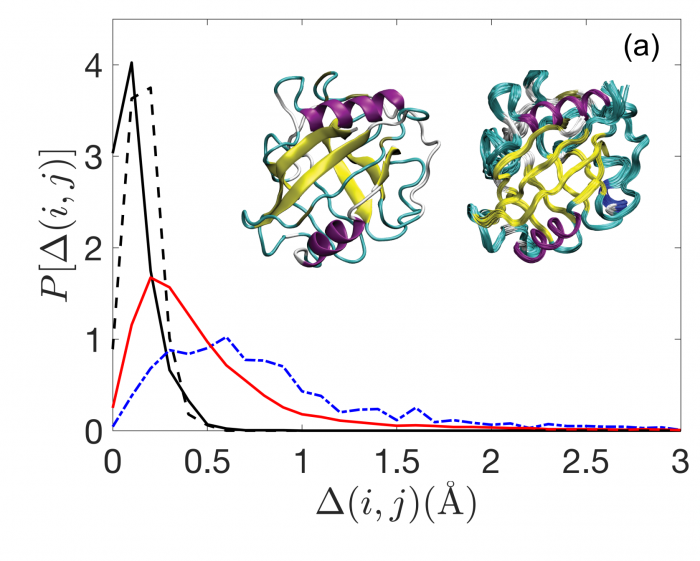
The team began by compiling a database of proteins with structures determined by both techniques at high resolution. That turns out to be a relatively small list—just 16 proteins fit the bill.
The researchers also created a database of x-ray crystallography protein structures determined by several different groups at different temperatures. This allowed the team to study how temperature influences structure.
They then created a mathematical model of the way proteins pack together to form either solid crystals or bundles in solution for NMR.
It turns out that the packing density can exactly explain the difference in structure for both techniques, the bundles in solution having a higher density than crystals. “We identify the physical basis for these differences by modelling protein cores as jammed packings of amino-acid-shaped particles,” the researchers say.
They can also fine-tune their mathematical model by varying the thermal energy used to generate the packings. Indeed, protein packing that is uninfluenced by temperature has about the same density as structures determined by x-ray crystallography
This suggests that temperature plays an important role in protein packing structure, since structures determined by NMR are denser. “These results indicate that thermalized systems can pack more densely than athermal systems, which suggests a physical basis for the structural differences between protein structures solved by NMR and x-ray crystallography,” say Mei and co.
However, temperature is not the whole story. Proteins in crystal structures are forced to take a certain shape, and this reduces the amount of thermal distortion the molecule can undergo.
So the Mei and co’s result raises an interesting question: To what extent is protein structure the result of temperature or the result of crystal packing?
That will have to wait for further work.
Ref: arxiv.org/abs/1907.08233 : Analyses Of Protein Cores Reveal Fundamental Differences Between Solution And Crystal Structures
Deep Dive
Biotechnology and health
How scientists traced a mysterious covid case back to six toilets
When wastewater surveillance turns into a hunt for a single infected individual, the ethics get tricky.
An AI-driven “factory of drugs” claims to have hit a big milestone
Insilico is part of a wave of companies betting on AI as the "next amazing revolution" in biology
The quest to legitimize longevity medicine
Longevity clinics offer a mix of services that largely cater to the wealthy. Now there’s a push to establish their work as a credible medical field.
There is a new most expensive drug in the world. Price tag: $4.25 million
But will the latest gene therapy suffer the curse of the costliest drug?
Stay connected
Get the latest updates from
MIT Technology Review
Discover special offers, top stories, upcoming events, and more.