An Algorithm Set To Revolutionize 3-D Protein Structure Discovery
One of the great challenges in molecular biology is to determine the three-dimensional structure of large biomolecules such as proteins. But this is a famously difficult and time-consuming task.
The standard technique is x-ray crystallography, which involves analyzing the x-ray diffraction pattern from a crystal of the molecule under investigation. That works well for molecules that form crystals easily.
But many proteins, perhaps most, do not form crystals easily. And even when they do, they often take on unnatural configurations that do not resemble their natural shape.
So finding another reliable way of determining the 3-D structure of large biomolecules would be a huge breakthrough. Today, Marcus Brubaker and a couple of pals at the University of Toronto in Canada say they have found a way to dramatically improve a 3-D imaging technique that has never quite matched the utility of x-ray crystallography.
The new technique is based on an imaging process called electron cryomicroscopy. This begins with a purified solution of the target molecule that is frozen into a thin film just a single molecule thick.
This film is then photographed using a process known as transmission electron microscopy—it is bombarded with electrons and those that pass through are recorded. Essentially, this produces two-dimensional “shadowgrams” of the molecules in the film. Researchers then pick out each shadowgram and use them to work out the three-dimensional structure of the target molecule.
This process is hard for a number of reasons. First, there is a huge amount of noise in each image so even the two-dimensional shadow is hard to make out. Second, there is no way of knowing the orientation of the molecule when the shadow was taken so determining the 3-D shape is a huge undertaking.
The standard approach to solving this problem is little more than guesswork. Dream up a potential 3-D structure for the molecule and then rotate it to see if it can generate all of the shadowgrams in the dataset. If not, change the structure, test it, and so on.
Obviously, this is a time-consuming process. The current state-of-the-art algorithm running on 300 cores takes two weeks to find the 3-D structure of a single molecule from a dataset of 200,000 images.
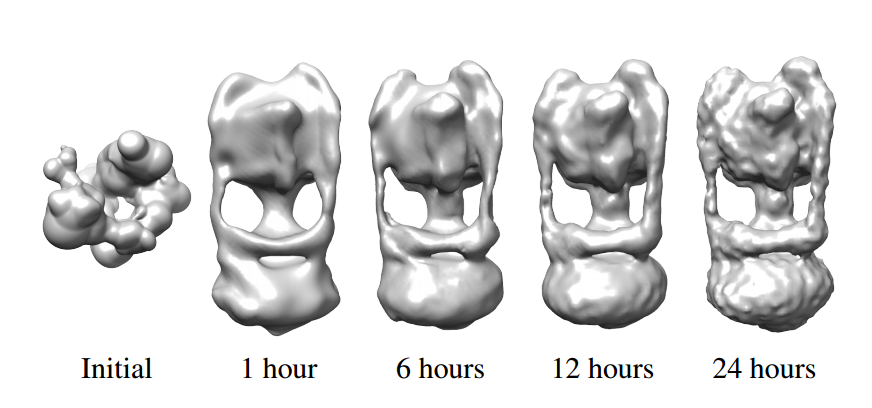
Brubaker and co have developed a much faster method that can do the same job in only 24 hours working on a single workstation. The technique relies on two algorithmic innovations.
The first exploits the fact that the images are noisy and so contain vast amounts of redundant information. The team gets around this using an algorithm that removes much of this redundancy, leaving only a subset of the original data. The trick, of course, is to get rid of the useless data while keeping the useful stuff, something they manage using a machine learning approach.
That reduces the amount of data that has to be processed but the main speed up comes from the second innovation, a statistical technique called importance sampling.
The main idea here is that certain pieces of data are more important than others in determining the final 3-D structure. So finding a way to focus on those can dramatically speed up the process.
Brubaker and co have found just such an approach. It turns out that large molecules frozen into thin films almost always end up lying on their side. So the shadowgrams nearly always show the molecules in this pose rather than standing on their head or bottom.
Building this knowledge into the algorithm dramatically increases the speed at which it settles on a potential 3-D structure since it can ignore the possibility that the images show the molecule from above or below.
The resulting improvement is huge. “This leads to speedups of 100,000-fold or more, allowing structures to be determined in a day on a modern workstation,” says Brubaker and co.
The team go on to demonstrate their technique on a set of shadowgrams of two well-known biomolecules. The first dataset consists of more than 46,000 images of a large transmembrane molecule called ATP synthase from the thermus thermophilus bacteria. The second consists of almost 6000 images of bovine mitochondrial ATP synthase.
The team also synthesized a third data set by taking 40,000 random shadowgrams of GroEL-GroES-(ADP)7, a biomolecule with a known structure. They then used their algorithm to work backward to recreate the original structure.
Finally, the team compares its approach to other state-of-the-art models and show that the new algorithm significantly outperforms these standard methods.
That is an impressive result that has the potential to dramatically change the landscape for molecular biologists who have struggled for years to find reliable new methods for determining the structure of large biomolecules.
Electron cryomicroscopy looks set to take on this role. And the technique is likely to get better as the resolution of this form of microscopy improves in the coming years.
Ref: arxiv.org/abs/1504.03573 : Building Proteins in a Day: Efficient 3-D Molecular Reconstruction
Keep Reading
Most Popular
Large language models can do jaw-dropping things. But nobody knows exactly why.
And that's a problem. Figuring it out is one of the biggest scientific puzzles of our time and a crucial step towards controlling more powerful future models.
How scientists traced a mysterious covid case back to six toilets
When wastewater surveillance turns into a hunt for a single infected individual, the ethics get tricky.
The problem with plug-in hybrids? Their drivers.
Plug-in hybrids are often sold as a transition to EVs, but new data from Europe shows we’re still underestimating the emissions they produce.
Stay connected
Get the latest updates from
MIT Technology Review
Discover special offers, top stories, upcoming events, and more.